
Molecular Modelling
Introduction
Each molecule in solution exhibits conformational preferences that depend strongly on the solvent type, the intrinsic structural properties of the investigated molecule and the existing boundaries. Understanding the change in this conformational space, which is often counterbalanced by direct interactions with the surface, is key to the formation of films at liquid-solid interfaces during the process of biomineralization or during the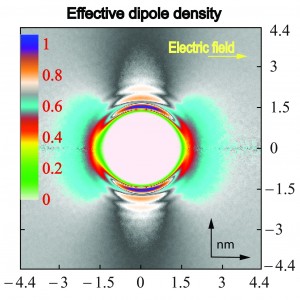 formation of technologically relevant, patterned surfaces. In the cellular context, “the boundaries” are provided by the cytoskeleton and the extracellular matrix. These are not only responsible for the structural integrity of living cells but are also part of an edifice that provides the working environment for the smaller, biochemically active, peptides and proteins, thereby determining the spatial coordination for the molecular recognition events. Consequently, biological signalling is subject to a plethora of physical constraints, where the material properties of the structure, as well as active and thermal fluctuations, affect the diffusion and the formation of macromolecular complexes.
formation of technologically relevant, patterned surfaces. In the cellular context, “the boundaries” are provided by the cytoskeleton and the extracellular matrix. These are not only responsible for the structural integrity of living cells but are also part of an edifice that provides the working environment for the smaller, biochemically active, peptides and proteins, thereby determining the spatial coordination for the molecular recognition events. Consequently, biological signalling is subject to a plethora of physical constraints, where the material properties of the structure, as well as active and thermal fluctuations, affect the diffusion and the formation of macromolecular complexes.
Our research
We are concerned with developing theoretical methods for de-convolving spectroscopic (CD, EPR, NMR, X-Ray) experiments which typically provide ensemble averaged information that cannot be understood without underlying modelling. This involves flexible molecules in bulk solutions, where, for example, the chiral nature of the solutes can affect their spectrum. Furthermore, we study specific and non-specific interactions of small molecules (peptides, complex ions) with solid surfaces, in the presence of complex solvents (water, ionic liquids). The nanometer to micron scales are investigated within advanced classical molecular dynamics approaches at various levels of coarse graining. Electronic transitions are obtained by ab initio and TD-DFT methods, which involve embedding into the classical environment in some of the available hybrid methods.
cannot be understood without underlying modelling. This involves flexible molecules in bulk solutions, where, for example, the chiral nature of the solutes can affect their spectrum. Furthermore, we study specific and non-specific interactions of small molecules (peptides, complex ions) with solid surfaces, in the presence of complex solvents (water, ionic liquids). The nanometer to micron scales are investigated within advanced classical molecular dynamics approaches at various levels of coarse graining. Electronic transitions are obtained by ab initio and TD-DFT methods, which involve embedding into the classical environment in some of the available hybrid methods.
Collaborations
The experimental insights in this project are provided by the groups of S. Horvat, D. Kralj (IRB, Zagreb, HR), A. Magerl (FAU, Erlangen, DE) and A. Rađjenović (EPFL, Lausanne). Successful exchange of the theoretical know-how involves the groups of D. M. Smith (IRB, Zagreb, HR) and S. J. Marrink (Groningen, NL).
People

The Application of the TDDFT Methods for the Calculation of the CD Spectra of Flexible Peptides
- We systematically tested performance of DFT functionals in calculations of CD spectra. As benchmark we used RICC2 calculations and the experimental findings.
Establishing conditions for simulating hydrophobic solutes in electric fields by molecular dynamics.
- We determine the conditions for stable performance of molecular dynamics simulations under electric field and provided an explanation for the long standing discrepancies in simulation results.
- We establish a method for calculating the spectra of a flexible peptide.
Protonation States of Histidines in the Active Site of (6-4) Photolyase.
- After showing that EPR experiments cannot be used uniquely to assign protonation states, we use pKA calculations and molecular dynamics to determine the states of relevant Histidines in the binding site.
Available projects
Funding